|
|
| |

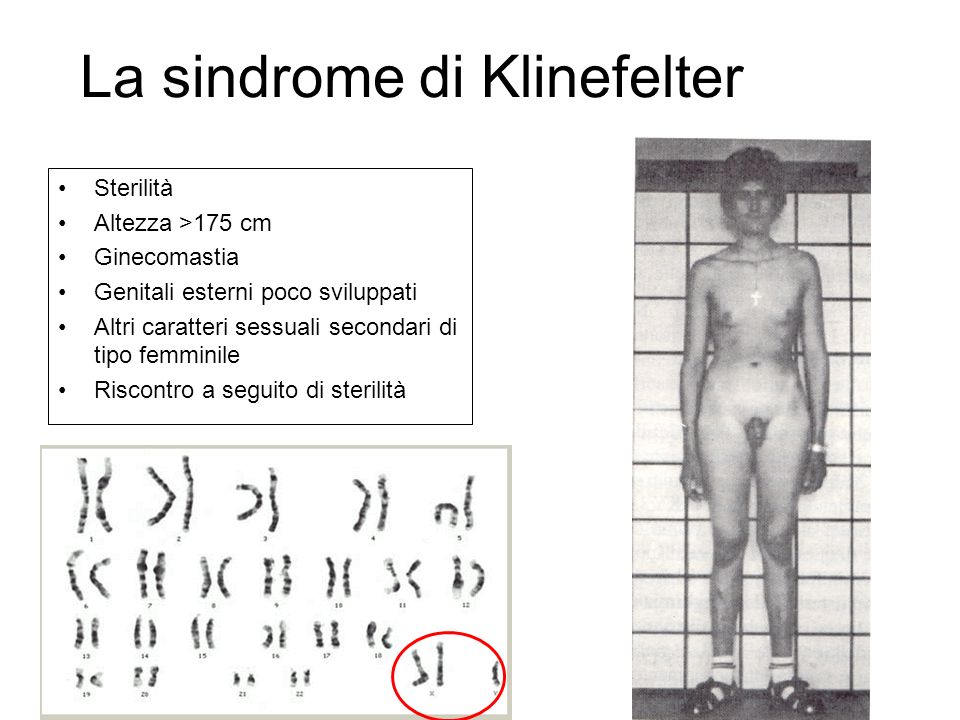
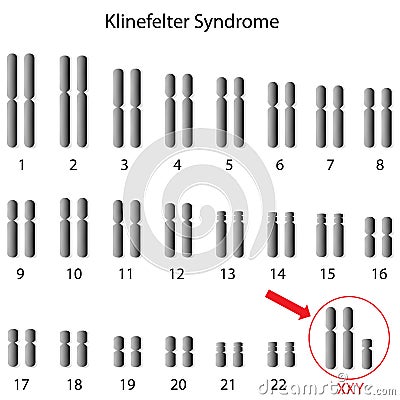
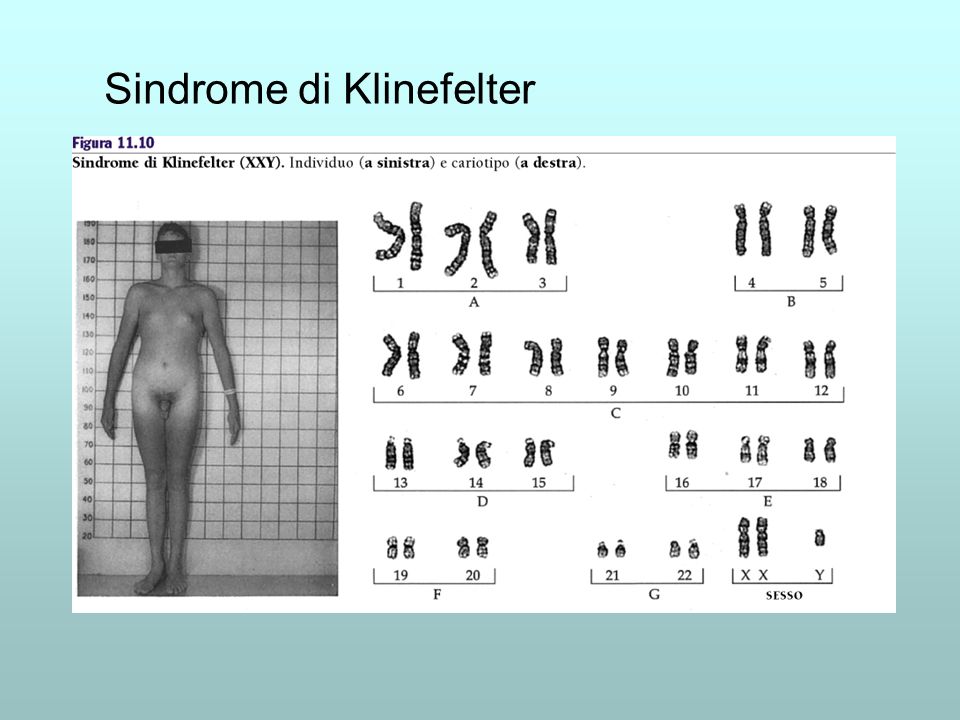
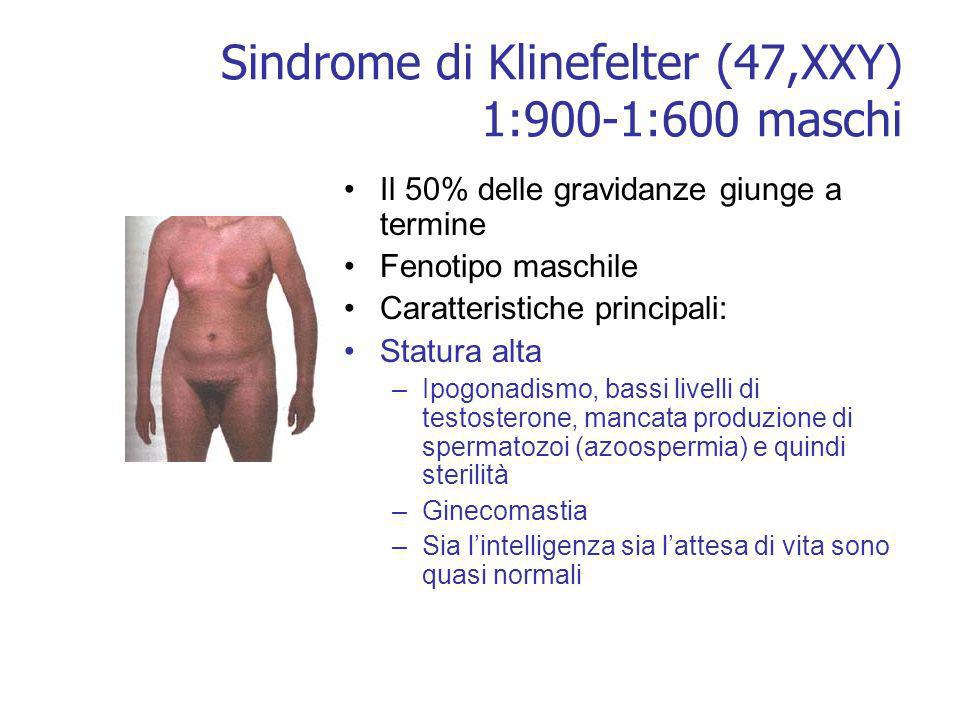
La sindrome di Klinefelter è una malattia genetica caratterizzata da un'anomalia cromosomica in cui un individuo di sesso maschile possiede un cromosoma X soprannumerario. Normalmente le donne possiedono due cromosomi sessuali XX e gli uomini uno X e uno Y: gli individui affetti dalla sindrome di Klinefelter hanno almeno due cromosomi X e almeno un cromosoma Y. Perciò gli individui con tale cariotipo sono solitamente indicati come "maschi XXY" o "47,XXY". È comunque presente un corpo di Barr, altrimenti assente nei maschi.
Questa condizione si verifica in circa 1-2 maschi su 1000 nati vivi. Molte persone affette dalla sindrome di Klinefelter non presentano alcun segno fino alla pubertà, quando le caratteristiche fisiche della malattia diventano più evidenti; in alcuni casi non si verifica una sintomatologia conclamata, con l'eccezione della sterilità o comunque di una riduzione della fertilità, e la diagnosi è conseguentemente formulata solo in età adulta.
Nella popolazione umana, la condizione 47, XXY è la più comune aneuploidia dei cromosomi sessuali nei maschi. Anche in altri mammiferi, come ad esempio i topi, possono verificarsi casi di sindrome XXY. Circa l'80% dei soggetti con Klinefelter possiede un cariotipo 47, XXY, mentre nel rimanente 20% dei casi si includono aneuploidie maggiori, mosaici 47, XXY/46, XY e anomalie strutturali del cromosoma X.
Le manifestazioni principali includono l'ipogonadismo e la riduzione della fertilità. Sono comuni, inoltre, altre differenze fisiche e comportamentali, anche se la loro gravità varia da individuo a individuo. In genere gli individui affetti da sindrome di Klinefelter tendono all'obesità.
Cenni storici
La sindrome prende il nome da Harry Klinefelter, che la descrisse per primo nel 1942, lavorando con Fuller Albright presso il Massachusetts General Hospital di Boston. Prima che il nome "sindrome di Klinefelter" entrasse nel linguaggio medico comune, la condizione veniva identificata come appartenente alle "disgenesie dei tubuli seminiferi".
Epidemiologia
La sindrome, diffusa equamente in tutte le etnie, è il più comune disordine genetico legato agli eterosomi, con prevalenza di 1-2 su 1000 maschi nella popolazione generale. Il 3,1% dei maschi infertili ne sono affetti e, inoltre, la sindrome è la causa principale di ipogonadismo maschile. Secondo una metanalisi la prevalenza della sindrome di Klinefelter è aumentata negli ultimi decenni; questo dato non sembra però essere correlato con l'aumento dell'età media della madre al concepimento, in quanto non è stato osservato alcun aumento nella prevalenza delle altre trisomie dei cromosomi sessuali (XXX e XYY).
Eziologia
Un individuo umano è caratterizzato fisiologicamente da due cromosomi sessuali. Un maschio con cariotipo normale presenta un cromosoma X e un cromosoma Y, con quest'ultimo che determina il sesso maschile; una donna al contrario due cromosomi X. Il fattore causale della sindrome di Klinefelter è la presenza di almeno un cromosoma X soprannumerario, che altera i livelli ormonali specifici dei maschi legati alla fisiologia sessuale, agendo come se fosse il secondo cromosoma X femminile, in particolare riducendo i valori sierici del testosterone. Un maschio affetto da tale patologia possiede quindi una configurazione genetica XXY che presenta sia la coppia normale di cromosomi maschile XY, sia quella femminile XX.
Si ritiene che la presenza di un cromosoma X soprannumerario sia causata da un evento di non disgiunzione durante la meiosi, che può avvenire da parte sia materna, sia paterna. Non vi sono fattori protettivi per evitare che ciò accada.
Nel caso in cui il cromosoma X in più provenga da parte paterna, l'evento ha origine durante la meiosi I (gametogenesi). La non disgiunzione si verifica quando i cromosomi omologhi, in questo caso X e Y, non riescono a separarsi, come dovrebbe accadere nella spermatogenesi, producendo uno spermatozoo con due cromosomi: uno X e uno Y. La successiva fecondazione di un normale uovo femminile (X) produce una prole di tipo XXY. La disposizione del cromosoma XXY è una delle più comuni variazioni genetiche del cariotipo XY e si verifica in circa 1 su 500 maschi nati vivi.
Diversamente, se il cromosoma soprannumerario proviene da parte materna, la non disgiunzione avviene durante la meiosi II. Ciò si verifica quando i cromatidi fratelli in un cromosoma sessuale femminile, in questo caso uno dei due X, non riescono a separarsi. Di conseguenza viene a crearsi un uovo XX che, una volta fecondato con uno spermatozoo Y, genera una prole XXY. Si ritiene che la trasmissione per via materna sia più frequente di quella paterna. L'età avanzata della madre è un fattore predisponente, anche se in misura molto limitata e nettamente inferiore al peso che può avere per la sindrome di Down.
Nei mammiferi con più di un cromosoma X, i geni di uno dei due cromosomi X non vengono espressi: questo fenomeno è noto come inattivazione del cromosoma X. Questo accade nei maschi XXY così come nelle normali femmine XX. Tuttavia, nei maschi XXY, alcuni geni situati nelle regioni pseudoautosomiche dei cromosomi X hanno geni omologhi corrispondenti sul cromosoma Y e sono dunque in grado di essere espressi.
La prima descrizione di un caso di un uomo con cariotipo 47, XXY, pubblicata nel 1959, fu ad opera di Patricia Jacobs e di John Strong del Western General Hospital di Edimburgo, in Scozia. Questo cariotipo è stato trovato in un maschio di 24 anni che presentava i tipici segni della sindrome di Klinefelter.
Varianti
Tutte le forme di sindrome di Klinefelter sono caratterizzate dalla presenza di almeno un cromosoma X soprannumerario in un fenotipo maschile. Vi sono però alcune varianti.
Il cariotipo 48, XXYY si verifica in 1 caso ogni 18.000-40.000 nascite maschili. Questo fenotipo non differisce molto da quello più comune (47, XXY) se non per la maggior altezza media riscontrata in età adulta. La variante 49, XXXXY è una rara polisomia che si riscontra in circa un caso ogni 85.000 bambini maschi nati. Questa condizione viene generalmente riconosciuta in età precoce per via dei gravi deficit che comporta, tra cui: marcato ritardo mentale, dismorfismi facciali (collo corto, occhi distanziati, bocca e naso larghi, strabismo e grandi orecchie), criptorchidismo, genitali ambigui e difetti scheletrici (cifosi, scoliosi, coxa valga) e cardiaci. Il primo paziente affetto da questa variante fu descritto nel 1960 e da allora al 2012 sono stati riportati in letteratura poco più di 100 casi.
In letteratura sono stati riportati anche casi molto più rari di cariotipo 48, XXXY e 48, XXYY caratterizzati da quadri fenotipici meno gravi rispetto al 49, XXXXY ma più severi rispetto al cariotipo classico: si presentano malformazioni fisiche, deficit di apprendimento e disturbi psicologici. Sono stati documentati anche dei rarissimi casi di varianti che comprendono i cariotipi: 49, XXXYY, 48, XYYY, 49, XYYYY, e 49, XXYY. Tutti comportano aspetto dismorfico e forte ritardo mentale.
I maschi con sindrome di Klinefelter possono presentare un mosaicismo nel cariotipo di forma 47, XXY/46, XY che comporta vari gradi di insufficiente spermatogenesi. Il mosaicismo 47, XXY/46, XX aggiunge al fenotipo le altre caratteristiche cliniche della sindrome, ma si riscontra molto raramente e, al 2006, solo 10 casi sono stati descritti.
Genetica
Come negli individui di sesso femminile, il cromosoma X soprannumerario viene inattivato in maniera casuale, in un fenomeno detto lyonizzazione. È possibile che l'inattivazione in una cellula staminale venga poi ereditata dalla popolazione di cellule figlie, ma nel complesso l'individuo è un mosaico potendo avere, in linea di massima, metà delle cellule con inattivazione di un cromosoma X e metà con l'inattivazione dell'altro cromosoma X. In tal senso, tutti gli studi volti a valutare la correlazione dei sintomi con una determinata origine parentale del cromosoma X sono inconsistenti.
Il gene deputato a tale processo è XIST (X inactive specific transcript), che trascrive un RNA espresso solo dal cromosoma inattivato e che non codifica per alcuna proteina. Agisce sul centro di inattivazione dell'X (Xic, X inactivation center) e non è espresso nel maschio con cariotipo normale (46, XY). A priori, ognuno dei due cromosomi X dei maschi con sindrome di Klinefelter hanno la medesima probabilità di venire attivati, ma non tutti i geni vengono silenziati: il 15% rimane in duplice copia biallelica e paiono questi i primi responsabili del fenotipo della sindrome.
Il cromosoma X differisce dagli autosomi per alcune caratteristiche relative ai geni, che sono corti, in numero minore e meno densamente presenti. Questi sono caratterizzati inoltre da un alto grado di conservazione, ma non del loro ordine. Il pattern polisomico, cioè il numero di cromosomi X presenti nel cariotipo dell'individuo (45, X0, 47, XXY, 47, XXX, 48, XXXX, e via dicendo), correla fortemente con la sintomatologia associata. Ciò ha fatto ipotizzare[37] un attivo ruolo del cromosoma X non solo nella determinazione del sesso, ma anche nello sviluppo neurologico e nelle funzioni cerebrali e comportamentali. Il dato è stato confermato da numerosi studi[38][39][40][41][42][43][44][45][46]. I geni del cromosoma X vengono infatti espressi non solo durante le prime fasi della spermatogenesi, ma anche nei muscoli scheletrici, negli ovari, nella placenta e nel cervello.
Nel locus Xq11-12 ha sede il gene RA, che codifica per il recettore degli androgeni, un recettore nucleare dotato di un dominio che riconosce selettivamente gli androgeni. Tale dominio è codificato da un tratto altamente polimorfico del primo esone, in cui è presente una sequenza ricca in triplette CAG. Proteine con un'espansione CAG corta sono molto affini e sensibili agli androgeni, viceversa, una lunga espansione CAG è poco affine e, se la ripetizione supera il numero di 40, insorge una grave malattia, l'atrofia spinobulbare di Kennedy, con interessamento neuromuscolare, marcato ipogonadismo primario e ginecomastia di grado variabile. Nei maschi con sindrome di Klinefelter viene inattivato il gene RA con l'espansione CAG più corta, cioè l'RA più attivo. I soggetti quindi non solo possiedono una minore produzione di androgeni, ma hanno anche un recettore meno affine.
La lunghezza dell'espansione CAG è finora l'unico parametro geneticamente individuabile che correla direttamente con l'ampio grado di variabilità fenotipica della sindrome di Klinefelter.
Clinica
Lo spettro delle manifestazioni cliniche è molto ampio: la malattia può presentarsi con un assetto testicolare di tipo fetale, deficit di androgeni, testicoli e pene piccoli. Durante l'età adulta può esserci azoospermia. I testicoli tendono ad essere piccoli a tutte le età per l'ipoplasia sia delle cellule germinali, sia delle cellule interstiziali. Infine, negli individui affetti da sindrome di Klinefelter è frequente la ginecomastia.
Diversamente da altre sindromi da polisomia del cromosoma X che presentano ritardo mentale con una prevalenza maggiore, nella sindrome di Klinefelter solo il 10% dei soggetti presenta un ritardo mentale. I problemi cognitivi sono meno pervasivi e più selettivi. Sul piano neurologico, la sindrome di Klinefelter è associata a ridotto sviluppo del linguaggio, con problemi di espressività, anomia, disartria. Sul piano comportamentale si possono riscontrare immaturità, poca sicurezza, timidezza.
Sviluppo neurologico e comportamentale
L'intelligenza generale è misurata mediante il FSIQ (full scale IQ), che nei maschi Klinefelter risulta nei limiti di normalità. Se, infatti, il QI nei bambini e adolescenti affetti da Klinefelter tende a essere più basso rispetto ai gruppi di riferimento, nell'età adulta può non esserci alcuna disparità. Vi è invece spesso discrepanza tra il QI verbale e il QI di performance. Questa discrepanza è stata osservata sia nei bambini, sia negli adulti affetti.
Gli studi suggeriscono che il difetto principale nei ragazzi è verbale, e appare evidente durante il periodo scolare. A 7 anni l'individuo ha da moderati a gravi problemi con la lettura, con l'articolazione delle parole, con la scrittura, mentre i problemi riguardanti la matematica intervengono poco più tardi. Approssimativamente il 50-75% dei ragazzi con Klinefelter dimostra una certa difficoltà d'apprendimento e di questi il 60-86% richiede una speciale educazione.
Gli adolescenti affetti mostrano una ridotta fiducia in loro stessi, sono riservati, hanno difficoltà a contenere l'impulsività e ad accettare le regole. Questi sintomi hanno implicazioni a lungo termine sulla socialità e sulle attività scolastiche e possono persistere nell'età adulta. Il maladattamento però non significa un disadattamento. L'impegno nella scuola aiuta spesso il giovane a superare questi problemi comportamentali.
Altre manifestazioni cliniche e fisiche
Altre possibili caratteristiche somatiche sono una ridotta circonferenza cranica, scarsi peli sul corpo, spalle strette e fianchi larghi, scarsa muscolatura. Sintomi addizionali includono l'aumentato rischio di osteoporosi, disordini autoimmuni della tiroide e diabete mellito di tipo 2. Vi è un rischio aumentato del 69% di venir ricoverati in ospedale anche prima della diagnosi di Klinefelter. Alcuni studi hanno associato la sindrome a un'altezza leggermente maggiore della media e a una predisposizione al sovrappeso.
I pazienti affetti da sindrome di Klinefelter vanno incontro a particolari alterazioni ormonali. I valori sierici dell'ormone follicolo-stimolante, dell'ormone luteinizzante, dell'ormone antimulleriano e dell'inibina B risultano normali nell'età prepuberale, mentre diventano anomali col passare del tempo. Uno studio condotto su individui adulti affetti dalla sindrome, ha rilevato livelli di testosterone bassi nel 45% dei casi e il 43,6% dei pazienti accusava disfunzioni sessuali.
La probabilità di sviluppare tumori è peculiare, in quanto alcune malattie sono strettamente correlate, come i tumori primitivi a cellule germinali del mediastino e il cancro della mammella, mentre per altre la sindrome pare essere un fattore di protezione, come nel caso del cancro della prostata. Utilizzando come parametro statistico l'eccesso di rischio assoluto (AER, absolute excess risk) per 100.000 soggetti l'anno, risulta aumentata la mortalità per cancro ai polmoni (AER +23,7), linfomi non-Hodgkin (AER +12,1) e tumore alla mammella maschile (AER +9,3).
L'assetto 48, XXXY è associato a collo corto, pieghe epicantiche, clinodattilia, sinostosi radio-ulnare e ritardo mentale di grado da moderato a severo. Associata al cariotipo 49, XXXXY si riscontra un'aumentata incidenza di anomalie cardiache congenite, in particolar modo la pervietà del dotto arterioso di Botallo. Il ritardo mentale è spesso grave. Le anomalie scheletriche includono sinostosi radio-ulnare, ginocchio valgo, petto escavato e clinodattilia.
Fertilità
In circa il 3,1% di maschi azoospermici viene riscontrato il cariotipo 47, XXY. La maggior parte dei maschi affetti è azoospermica e l'esecuzione di una biopsia testicolare può rivelare un'assenza di cellule della linea germinale, ipertrofia delle cellule di Leydig e una marcata fibrosi dei tubuli seminiferi. Inoltre si ricorda che la terapia androgenica, usata nel trattamento della sindrome di Klinefelter, influenza negativamente la fertilità.
Sono state descritte gravidanze da concepimento naturale con partner affetti da sindrome di Klinefelter e ciò è più frequente nei casi di mosaicismo. Nei casi di spermatogenesi residua presente, le tecniche di fecondazione assistita possono dare loro la possibilità di avere figli propri, ma poiché la tecnica bioptica di estrazione degli spermatozoi (testicular sperm extraction o TESE) è invasiva, occorre sempre valutare prima la presenza di alcuni indicatori che suggeriscono la possibile presenza di una residua spermatogenesi. Il riscontro di spermatogoni e spermatozoi è più frequente nel giovane, per esempio, perché il numero di gameti diminuisce rapidamente con l'età, ma la somministrazione di inibitori dell'aromatasi e di gonadotropina corionica umana, per stimolare la produzione endogena testicolare di testosterone, fa aumentare la conta spermatica. Le frazioni di spermatozoi così ottenute dimostrano la presenza di gameti anormali, non euploidi, dal 7 al 20% (nel maschio 46, XY tale quota è inferiore all'1%).
Diagnosi
Circa il 10% dei casi di Klinefelter ha diagnosi prenatale[89]. Le manifestazioni cliniche iniziali possono comparire nella prima infanzia o, più spesso, durante la pubertà e comprendono il mancato sviluppo dei caratteri sessuali secondari, la microrchidia e l'aspermatogenesi. La tendenza all'alta statura è difficilmente diagnosticabile durante la pubertà[54]. Nonostante la presenza di testicoli piccoli, solo un quarto dei maschi affetti è riconosciuto durante la pubertà, mentre il 25% dei casi viene diagnosticato tardivamente in età adulta. Si stima che circa il 64% degli individui affetti non vengano riconosciuti come tali. Non vi sono chiari riferimenti per sospettare la sindrome di Klinefelter: spesso la diagnosi avviene accidentalmente in seguito ad esami e visite mediche per motivi non strettamente legati alla condizione.
L'analisi del cariogramma sui linfociti è lo standard genetico per effettuare una diagnosi. In passato era pratica comune anche l'osservazione del corpo di Barr. Per confermare il mosaicismo viene effettuata l'analisi del cariotipo anche su fibroblasti cutanei o tessuto testicolare.
Altre metodiche diagnostiche comprendono la ricerca di elevati livelli sierici di gonadotropine (ormoni FSH e LH), la presenza di azoospermia e la determinazione della cromatina sessuale in tamponi orali. Uno studio del 1994 ha proposto, come alternativa, l'uso della reazione polimerasica a catena (PCR) come metodo diagnostico più veloce. L'esame è positivo se rivela la presenza di RNA che porta le informazioni di un gene, contenuto nel cromosoma X, che serve come un marcatore per l'inattivazione del secondo (e di eventuali ulteriori) X supplementare. Questo gene, chiamato X-inactive-specific transcript (XIST), è trascritto solamente nel cromosoma X inattivo.
Diagnosi differenziale
La sindrome di Klinefelter entra in diagnosi differenziale con altre due condizioni genetiche: la sindrome dell'X fragile (causata da una mutazione del gene FMR1 sul cromosoma X) e la sindrome di Marfan (una malattia autosomica dominante che colpisce il tessuto connettivo). La causa dell'ipogonadismo, proprio della sindrome, può essere attribuita a molte altre diverse condizioni mediche.
Sono stati documentati rari casi di individui affetti da sindrome di Down che presentavano anche il difetto 47/XXY.
Trattamento
La mutazione genetica è irreversibile, tuttavia, per gli individui affetti che desiderano avere un aspetto più maschile, è possibile ricorrere alla somministrazione di testosterone. Uno studio, effettuato su pazienti adolescenti trattati con impianto sottocutaneo a rilascio controllato di tale ormone, ha dimostrato buoni risultati malgrado la necessità di un costante monitoraggio. La terapia ormonale è utile anche per prevenire l'insorgenza dell'osteoporosi.
Spesso i maschi che presentano ginecomastia e/o ipogonadismo soffrono di depressione e/o ansia sociale. Almeno uno studio consiglia l'opportunità di un sostegno psicologico per i giovani affetti da sindrome di Klinefelter al fine di ridurre i loro deficit psicosociali. L'intervento chirurgico di mastectomia può essere preso in considerazione sia per i problemi psicologici dovuti alla ginecomastia, sia per ridurre la probabilità di sviluppare un tumore mammario.
Il ricorso ad una terapia comportamentale può mitigare gli eventuali disturbi del linguaggio, le difficoltà scolastiche e di socializzazione. Un approccio mediante terapia occupazionale è utile nei bambini affetti dalla sindrome che presentano disprassia motoria.
Trattamento contro l'infertilità
Fino al 1996, gli uomini che presentavano un cariotipo tipico della sindrome di Klinefelter erano considerati generalmente sterili. Tuttavia, al 2010, sono state documentate più di 100 gravidanze di successo realizzate tramite la tecnica FIVET con spermatozoi prelevati chirurgicamente da uomini con la sindrome (testicular sperm extraction o TESE).
Uno studio effettuato sui risultati di 54 TESE ha evidenziato che il tasso di recupero degli spermatozoi è del 72% per ogni procedura e che il 69% degli uomini possedevano un numero adeguato di spermatozoi per realizzare l'iniezione intracitoplasmatica dello spermatozoo. Il 46% delle gravidanze ottenute in questo modo si sono concluse positivamente e tutti i bambini nati presentavano un cariotipo normale. Circa il 30% dei pazienti che si sottopongono a questa procedura ottiene questi risultati.
Talvolta viene, inoltre, eseguita la crioconservazione del seme prelevato in età adolescenziale per poter tentare un'eventuale futura procreazione.
Prognosi
I bambini con la forma XXY differiscono poco dai bambini sani. Nonostante possano dover affrontare durante l'adolescenza problemi spesso emotivi e comportamentali e avere difficoltà nei risultati scolastici, la maggior parte di loro può raggiungere una piena indipendenza dalle loro famiglie in età adulta. Alcuni riescono ad arrivare ad avere un'istruzione universitaria e una vita pressoché normale.
I risultati di uno studio effettuato su 87 adulti australiani con sindrome di Klinefelter evidenzia come coloro che hanno avuto una diagnosi e un adeguato trattamento fin dalla giovane età ne abbiano tratto un significativo beneficio rispetto a chi ha ricevuto la diagnosi in età adulta.
Non sembra esserci una sostanziale diminuzione dell'aspettativa di vita da parte degli individui con sindrome di Klinefelter. Diversi studi sono stati compiuti e hanno portato a risultati non definitivi. Un primo lavoro edito nel 1985 individuava una maggior mortalità dovuta prevalentemente a valvulopatie aortiche, allo sviluppo di tumori e a possibili emorragie subaracnoidee, tale da ridurre di circa 5 anni l'aspettativa di vita. Studi successivi hanno ridotto questa stima, associando alla condizione una riduzione mediana della sopravvivenza di 2,1 anni. Tuttavia questi dati non sono assoluti e hanno bisogno di ulteriori verifiche.
A seguito alla diagnosi della sindrome non si rende necessario nessun tipo di follow up
Wiki
|
|

 .
.
 .
.
 Grafica Magica Tutorial Forum - Photoshop, Gimp e Paint Shop Pro
Grafica Magica Tutorial Forum - Photoshop, Gimp e Paint Shop Pro 
 Contacts
Contacts Web
Web